Ligandbook contains parameter sets for Aspirin for the OPLS-AA force-field under LigandBook ID LBID 785 at
https://ligandbook.org/package/permalink/785/latest
However the atom order will be different from what's available in the PDB sturcutures of aspirin-protein complexes. This makes using these parameters hard – one has to manually re-order the atoms, something that's error-prone and tedious. Ligandbook has a function that allows parameter files to be re-ordered to match your input PDB file.
We want to simulate the system basing on the following 1OXR PDB file.
Aspirin induces its Anti-inflammatory effects through its specific binding to Phospholipase A2: Crystal structure of the complex formed between Phospholipase A2 and Aspirin at 1.9A resolution DOI: 10.2210/pdb1oxr/pdb
Let's start by downloading the PDB file
$ wget http://files.rcsb.org/view/1oxr.pdb
Select out the ligand (resname AIN) using a text editor, VMD or, as we do in the following, MDAnalysis.
#!/usr/bin/env python
from MDAnalysis import Universe
u = Universe("1oxr.pdb")
u.select_atoms("resname AIN").write("1oxr_resname-AIN.pdb")
This writes out a ligand-only file (no hydrogens)
To protonate the ligand, we've used Schrodinger's Maestro PrepWizard and exported the protonated ligand (charged, -1 form) as 1oxr_resname-AIN_Hs.pdb. Alternatively, you could use RDKit (free and open source).
On the main Ligandbook page, upload the 1oxr_resname-AIN.pdb or 1oxr_resname-AIN_Hs.pdb to get the re-orderd parameter files.
If you haven't protonated the ligand manually, Ligandbook will generate re-ordered parameters for all protonation states of the molecule in the database.
If the upload and re-ordering is successful you should see the following page:
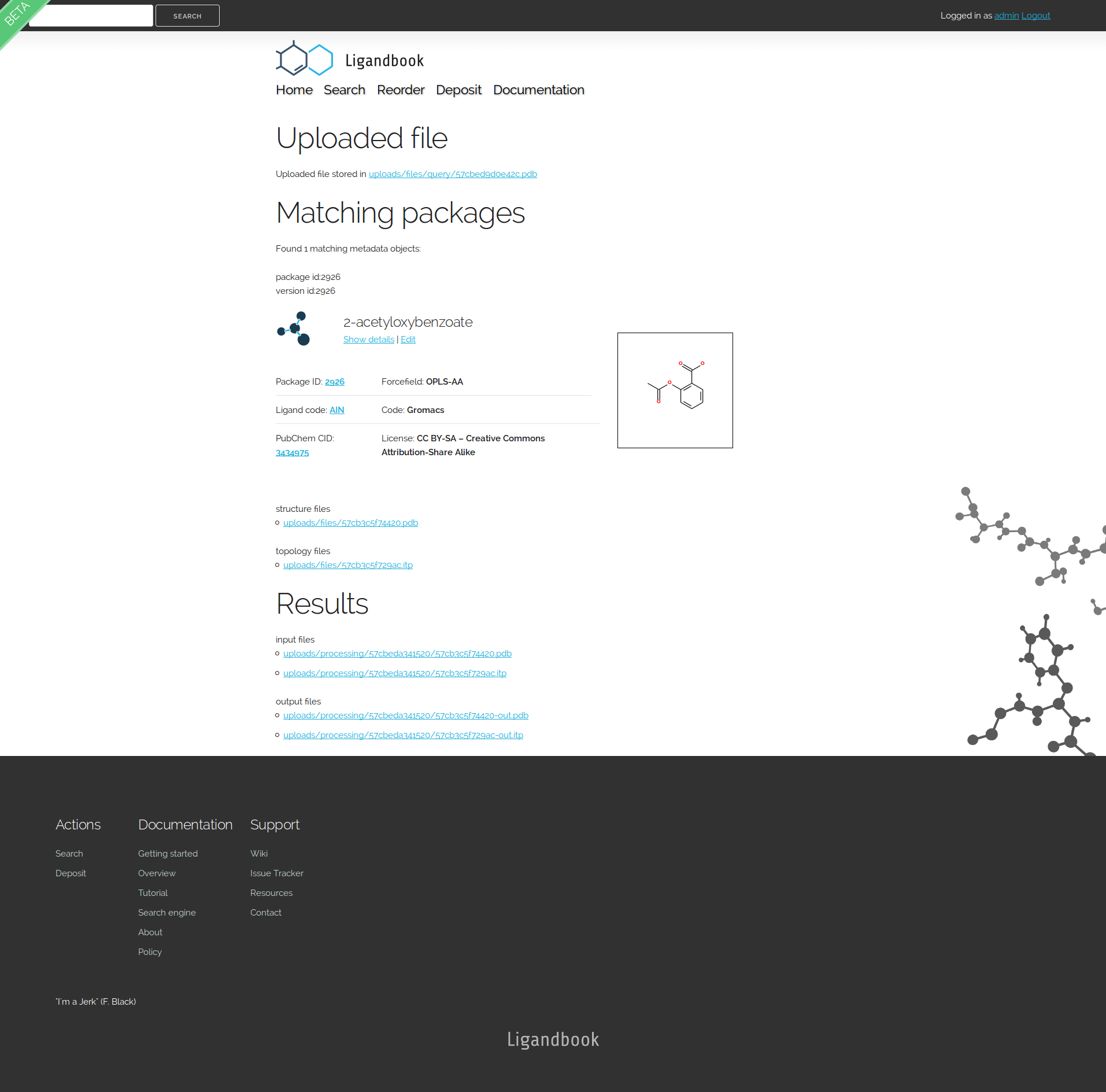
The charged aspirin package has structure and topology files called '57b9d65c162e4.pdb' and '57b9d65c14721.itp' respectively, so we're going to download '57b9d65c162e4-out.pdb' '57b9d65c14721-out.itp' from the Results section.
From the PDB file select only the protein and calcium ion.
#!/usr/bin/env python
from MDAnalysis import Universe
u = Universe("1oxr.pdb")
u.select_atoms("resname CA").set_segids("B")
u.select_atoms("protein or resname CA").write("protein.pdb")
Generate separate topologies for the protein Ca2+ ion
gmx pdb2gmx -f protein.pdb -o protein.gro -merge no 15: OPLS-AA/L all-atom force field (2001 aminoacid dihedrals) 3: TIP3P TIP 3-point
Manually modify topol.top to include '57b9d65c14721-out.itp' and add AIN to '[ molecules ]' section.
; Include topology for ions #include "oplsaa.ff/ions.itp" ; Include topology for ligand #include "57b9d65c14721-out.itp" [ system ] ; Name PHOSPHOLIPASE A2 ISOFORM 3; [ molecules ] ; Compound #mols Protein_chain_A 1 Ion_chain_B 1 AIN 1
Merge the protein and ion structure with the ligand structure. Confirm visually in VMD that the ligand is at the right position.
#!/usr/bin/env python
from MDAnalysis import Universe, Merge
p = Universe("protein.gro")
l = Universe("57b9d65c162e4-out.pdb")
u = Merge(p.atoms, l.atoms)
u.atoms.write("conf.gro")
Finally adjust the box size to something reasonable, here a 5 nm cube.
gmx editconf -f conf.gro -o conf.gro -box 5 5 5
Download a simple MDP file for steepest-descent minimization minim.mdp.
Run grompp
gmx grompp -f minim.mdp Setting the LD random seed to 616040524 Generated 330891 of the 330891 non-bonded parameter combinations Generating 1-4 interactions: fudge = 0.5 Generated 330891 of the 330891 1-4 parameter combinations Excluding 3 bonded neighbours molecule type 'Protein_chain_A' Excluding 3 bonded neighbours molecule type 'Ion_chain_B' Excluding 3 bonded neighbours molecule type 'AIN' NOTE 2 [file topol.top, line 51]: System has non-zero total charge: -3.000000 Total charge should normally be an integer. See http://www.gromacs.org/Documentation/Floating_Point_Arithmetic for discussion on how close it should be to an integer. Removing all charge groups because cutoff-scheme=Verlet Analysing residue names: There are: 119 Protein residues There are: 1 Ion residues There are: 1 Other residues Analysing Protein... Analysing residues not classified as Protein/DNA/RNA/Water and splitting into groups... Analysing residues not classified as Protein/DNA/RNA/Water and splitting into groups... Number of degrees of freedom in T-Coupling group rest is 5250.00 Calculating fourier grid dimensions for X Y Z Using a fourier grid of 42x42x42, spacing 0.119 0.119 0.119 Estimate for the relative computational load of the PME mesh part: 0.50 NOTE 3 [file minim.mdp]: The optimal PME mesh load for parallel simulations is below 0.5 and for highly parallel simulations between 0.25 and 0.33, for higher performance, increase the cut-off and the PME grid spacing. This run will generate roughly 0 Mb of data There were 3 notes gcq#265: "set: No match." (tcsh)
Execute mdrun
gmx mdrun -v
Steepest Descents:
Tolerance (Fmax) = 1.00000e+03
Number of steps = 50000
Step= 0, Dmax= 1.0e-02 nm, Epot= -1.05985e+04 Fmax= 1.20986e+04, atom= 1728
Step= 1, Dmax= 1.0e-02 nm, Epot= -1.54148e+04 Fmax= 7.65660e+03, atom= 1729
Step= 2, Dmax= 1.2e-02 nm, Epot= -1.77683e+04 Fmax= 5.36818e+03, atom= 1729
Step= 3, Dmax= 1.4e-02 nm, Epot= -1.91571e+04 Fmax= 8.70649e+03, atom= 1736
Step= 4, Dmax= 1.7e-02 nm, Epot= -1.94310e+04 Fmax= 1.83734e+04, atom= 1736
Step= 5, Dmax= 2.1e-02 nm, Epot= -1.97775e+04 Fmax= 1.42154e+04, atom= 1736
Step= 7, Dmax= 1.2e-02 nm, Epot= -2.00253e+04 Fmax= 5.42548e+03, atom= 1736
Step= 8, Dmax= 1.5e-02 nm, Epot= -2.01336e+04 Fmax= 1.79265e+04, atom= 1736
Step= 9, Dmax= 1.8e-02 nm, Epot= -2.03538e+04 Fmax= 1.03227e+04, atom= 1736
Step= 11, Dmax= 1.1e-02 nm, Epot= -2.04666e+04 Fmax= 6.52568e+03, atom= 1736
Step= 12, Dmax= 1.3e-02 nm, Epot= -2.04942e+04 Fmax= 1.38291e+04, atom= 1736
Step= 13, Dmax= 1.5e-02 nm, Epot= -2.06030e+04 Fmax= 1.04612e+04, atom= 1736
Step= 15, Dmax= 9.3e-03 nm, Epot= -2.07028e+04 Fmax= 4.19987e+03, atom= 1736
Step= 16, Dmax= 1.1e-02 nm, Epot= -2.07199e+04 Fmax= 1.31600e+04, atom= 1736
Step= 17, Dmax= 1.3e-02 nm, Epot= -2.08254e+04 Fmax= 7.90087e+03, atom= 1736
Step= 19, Dmax= 8.0e-03 nm, Epot= -2.08845e+04 Fmax= 4.72093e+03, atom= 1736
Step= 20, Dmax= 9.6e-03 nm, Epot= -2.08968e+04 Fmax= 1.03994e+04, atom= 1736
Step= 21, Dmax= 1.2e-02 nm, Epot= -2.09576e+04 Fmax= 7.81277e+03, atom= 1736
Step= 23, Dmax= 6.9e-03 nm, Epot= -2.10126e+04 Fmax= 3.08803e+03, atom= 1736
Step= 24, Dmax= 8.3e-03 nm, Epot= -2.10267e+04 Fmax= 1.00567e+04, atom= 1736
Step= 25, Dmax= 1.0e-02 nm, Epot= -2.10907e+04 Fmax= 5.62295e+03, atom= 1736
Step= 27, Dmax= 6.0e-03 nm, Epot= -2.11245e+04 Fmax= 3.87609e+03, atom= 1736
Step= 28, Dmax= 7.2e-03 nm, Epot= -2.11399e+04 Fmax= 7.33757e+03, atom= 1736
Step= 29, Dmax= 8.6e-03 nm, Epot= -2.11719e+04 Fmax= 6.32620e+03, atom= 1736
Step= 31, Dmax= 5.2e-03 nm, Epot= -2.12090e+04 Fmax= 1.80368e+03, atom= 1736
Step= 32, Dmax= 6.2e-03 nm, Epot= -2.12349e+04 Fmax= 8.10774e+03, atom= 1736
Step= 33, Dmax= 7.5e-03 nm, Epot= -2.12807e+04 Fmax= 3.61644e+03, atom= 1736
Step= 35, Dmax= 4.5e-03 nm, Epot= -2.13013e+04 Fmax= 3.48689e+03, atom= 1736
Step= 36, Dmax= 5.4e-03 nm, Epot= -2.13179e+04 Fmax= 4.92513e+03, atom= 1736
Step= 37, Dmax= 6.4e-03 nm, Epot= -2.13355e+04 Fmax= 5.27419e+03, atom= 1736
Step= 38, Dmax= 7.7e-03 nm, Epot= -2.13449e+04 Fmax= 6.88341e+03, atom= 1736
Step= 39, Dmax= 9.3e-03 nm, Epot= -2.13562e+04 Fmax= 7.77583e+03, atom= 1736
Step= 41, Dmax= 5.6e-03 nm, Epot= -2.14004e+04 Fmax= 1.00781e+03, atom= 1736
Step= 42, Dmax= 6.7e-03 nm, Epot= -2.14333e+04 Fmax= 9.49746e+03, atom= 1736
Step= 43, Dmax= 8.0e-03 nm, Epot= -2.14894e+04 Fmax= 3.16515e+03, atom= 1736
Step= 45, Dmax= 4.8e-03 nm, Epot= -2.15007e+04 Fmax= 4.41849e+03, atom= 1736
Step= 46, Dmax= 5.8e-03 nm, Epot= -2.15134e+04 Fmax= 4.69975e+03, atom= 1736
Step= 47, Dmax= 6.9e-03 nm, Epot= -2.15192e+04 Fmax= 6.22042e+03, atom= 1736
Step= 48, Dmax= 8.3e-03 nm, Epot= -2.15278e+04 Fmax= 6.90746e+03, atom= 1736
Step= 50, Dmax= 5.0e-03 nm, Epot= -2.15621e+04 Fmax= 9.63041e+02, atom= 1736
writing lowest energy coordinates.
Steepest Descents converged to Fmax < 1000 in 51 steps
Potential Energy = -2.1562055e+04
Maximum force = 9.6304138e+02 on atom 1736
Norm of force = 8.1507614e+01
gcq#108: "Motherhood Means Mental Freeze" (The Breeders)
This is a minimal setup for an in-vacuo minimzation. It's a basic test case - if this flow described above works, you can proceed to adding water and ions to your system.
Jan Domański, Oliver Beckstein, Bogdan I. Iorga; Ligandbook: an online repository for small and drug-like molecule force field parameters, Bioinformatics, Volume 33, Issue 11, 1 June 2017, Pages 1747–1749, https://doi.org/10.1093/bioinformatics/btx037
"Whatever Happened to Pong ?" (F. Black)
